Epigenetic landscapes consist of distinct domains of euchromatin and heterochromatin, which are unique to different cell types and different stages in development. Epigenetic regulation of gene expression is mediated through posttranslational modifications of histones. For example, tri-methylation of histone H3 Lys4 is a modification associated with active chromatin (euchromatin), while tri-methylation of histone H3 Lys27 is a mark of inactive heterochromatin.
A powerful method used to identify localized regions of histone modifications as well as binding sites for transcription factors on a genome-wide scale is the chromatin immunoprecipitation (ChIP) assay. The ChIP assay uses formaldehyde to covalently crosslink proteins to their DNA substrates in living cells. The protein:DNA complexes are then immunoprecipitated using antibodies generated against specific histone modifications or transcription factors. The precipitated DNA is purified and can be analyzed by hybridization to oligo-nucleotide microarrays (ChIP-chip) or by high throughput DNA sequencing (ChIP-seq). These methods provide an opportunity to take a snapshot of DNA-protein interactions in a given cell type, using populations of cultured cells, subsets of cells taken at specific times of the cell cycle or development, or even cells taken directly from tissue samples. ChIP-chip was the first method used for whole genome binding site analyses. However, multiple DNA microarrays are required to cover the entire human genome, resulting in high costs for comprehensive studies. ChIP-seq technology is another method that offers the ability to identify binding sites across the entire genome in a single sequencing run. This can be done using the Illumina GA2 sequencer, which generates short sequence reads that are sufficient for accurate mapping of the enriched DNA fragments to their genomic location.
Using the SimpleChIP® Enzymatic Chromatin IP Kit (Magnetic Beads) #9003 from Cell Signaling Technology, we have performed ChIP-seq experiments to identify the epigenetic signatures of tri-methyl-histone H3 Lys4 and tri-methyl-histone H3 Lys27 histone marks in the K562 erythroleukemia cell line. Chromatin was prepared from K562 cells as described in the SimpleChIP® protocol and immunoprecipitated with Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit mAb #9751, Tri-Methyl-Histone H3 (Lys27) (C36B11) Rabbit mAb #9733 and Normal Rabbit IgG #2729 as a negative control. The enrichment of tri-methyl histone H3 Lys4 and tri-methyl histone H3 Lys27 at known binding sites was confirmed by standard polymerase chain reaction (Figure 1). The immunoprecipitated DNA was then used to prepare libraries for sequencing with the Illumina GA2 sequencing platform. Obtained sequences were mapped to UCSC Human Genome Assembly (HG18) and only uniquely mapped sequences were retained. Enriched binding sites are reflected by elevated number of sequence reads giving rise to peaks (Figure 2). The rabbit monoclonal antibodies were found to give excellent enrichment, as reflected in the average peak height of 17 sequence reads and very low background.

Figure 1. K562 chromatin was immunoprecipitated with Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit mAb #9751 (lane 1), Tri-Methyl-Histone H3 (Lys27) (C36B11) Rabbit mAb #9733 (lane 2) or Normal Rabbit IgG #2729 (lane 3), using the SimpleChIP® Enzymatic Chromatin IP Kit (Magnetic Beads) #9003. Purified DNA was analyzed by standard PCR methods using primers specific for the transcriptionally active DHFR gene and HIST1H2AC gene cluster, or the inactive MYT1 gene.
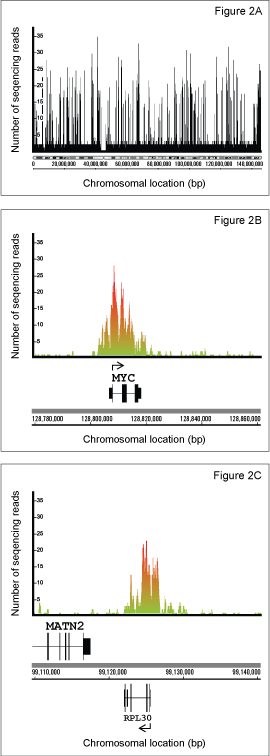
Figure 2. K562 chromatin was immunoprecipitated with Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit mAb #9751. The enriched DNA was sequenced using the Illumina GA2 Sequencer. The obtained sequences were mapped to the UCSC Human Genome Assembly (HG18) and data was visualized using IGB (Integrated Genome Browser). (Figure 2A) Enrichment of tri-methyl histone H3 Lys4 is shown for chromosome 8. Enriched sites of methylation are identified by the elevated number of sequencing reads for a given region of DNA. Enrichment of tri-methyl-histone H3 Lys4 methylation is shown for the active c-MYC (Figure 2B) and RPL30 (Figure 2C) genes.
CST would like to thank H. O'geen and P.J. Farnham of University of California, Davis in Davis, California for sharing their ChIP-Sequencing data.