
Overview of Metabolism and Metabolic Disorders
Metabolism and Metabolic Disorders
The word metabolism originates from the Greek “to change” and accordingly defines all of the chemical changes or processes within cells, tissues, and organisms that maintain homeostasis. The focus here is to review the different metabolic pathways at the cellular level and how these pathways, when perturbed, can contribute to different metabolic disorders. Being able to assess the multitude of signaling events and chemical reactions that underlie metabolic change is key to understanding normal cell physiology and disease. Having critical tools, such as antibodies specific to key enzymes and proteins involved in these processes, is vital to knowing how metabolism goes awry in human disease.
Cell Metabolism
Cellular metabolism is largely defined by the complex array of biochemical reactions involving biomolecular synthesis (anabolism), maintenance, or breakdown (catabolism), the sum total of which define the energetic status of the cell. Molecules involved in these metabolic processes include basic cellular building blocks such lipids, amino acids, carbohydrates and nucleotides, and numerous enzymes and cofactors that participate in the metabolic reactions. These numerous reactions are what drive cell metabolism and encompass every process that adds to or subtracts from the overall building block and energy pool of the cell. Nutrient uptake influences the main cellular metabolic reactions; thus, it's critical to understand the key nutrient metabolic systems in the cell, including lipid, carbohydrate, amino acid, and nucleotide metabolism.
Lipid Metabolism
Lipids, or fats, are important, energy-rich nutrients that cells use as fuel to support cell functions. In addition, lipids serve as important building blocks for key cellular structures (e.g., membranes), while also participating in many important signaling networks. Some cell types (e.g., adipocytes) are specialized for the storage of lipids, which can be mobilized through catabolism to support organismal function in conditions of nutritional or energetic deficiency. Key lipids, such as triglycerides and cholesterol, are obtained from the diet and have to be appropriately digested and absorbed.
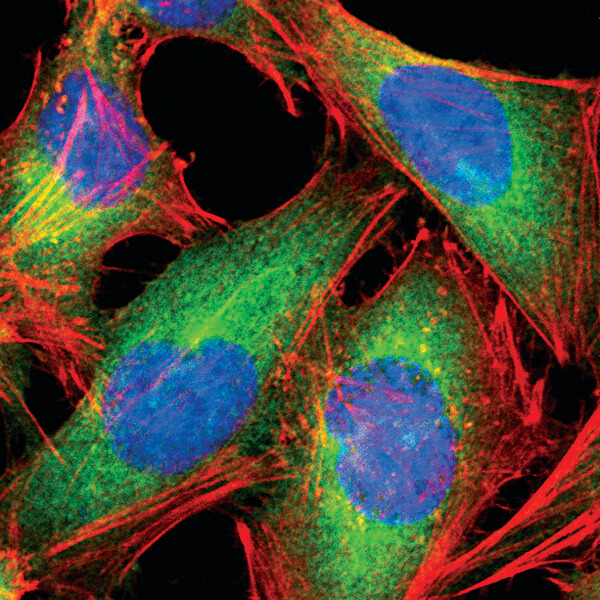
Fatty Acid Synthase (C20G5) Rabbit mAb #3180: Confocal IF analysis of HeLa cells using #3180 (green). Actin filaments were labeled with DY-554 phalloidin #13054 (red). Blue pseudocolor = DRAQ5 #4084 (fluorescent DNA dye).
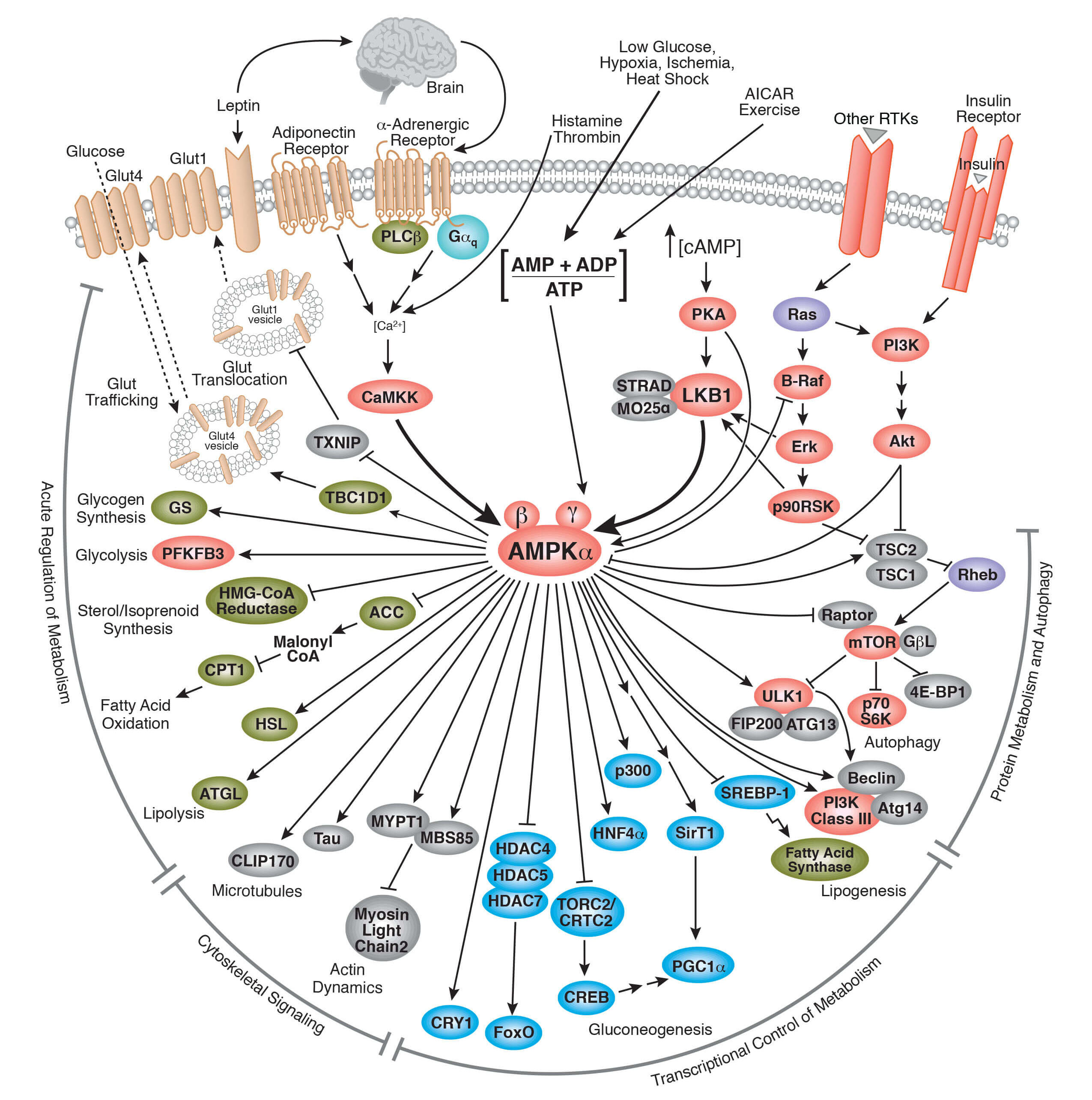
Carbohydrate Metabolism
Carbohydrates are "sugars" composed of carbon, hydrogen, and oxygen (CHO) that can exist as simple monomers (monosaccarides) such as glucose, or in more complex form, ranging from disaccharides (e.g., sucrose) to highly complex polysaccharides (e.g., starch). Carbohydrate metabolism begins immediately following food ingestion through the release of enzymes (e.g., salivary amylase) which initiate the breakdown of polysaccharides into less complex sugars. Digestion continues in the small intestine, where pancreatic amylase completes the breakdown of polysaccharides into monosaccharides. Glucose is the primary monosaccharide in the human diet, providing for much of the daily energetic needs of an individual.
Carbohydrate metabolism begins in the mouth with an enzyme known as salivary amylase. Salivary amylase breaks down complex carbohydrates (i.e., polysaccharides) into less complex molecules. Digestion of carbohydrates continues in the small intestine where pancreatic amylase further breaks down the partially digested polysaccharides into their simplest form (i.e., monosaccharides). Glucose is the most essential monosaccharide since it provides the majority of the fuel needed by body. Glucose is absorbed into the blood stream in the small intestine and transported by the circulatory system to all organs, where its uptake into cells is induced via insulin. Glucose uptake is particularly prominent in liver and muscle cells, where metabolic enzymes convert glucose into the polysaccharide glycogen through the process of glycogenesis. Glycogenesis serves an important energy storage function, as glycogen stores can be rapidly hydrolyzed back into glucose, providing the body with a readily available source of energy (glucose) if blood glucose levels drop. Glycogen is composed of multiple glucose subunits, acts as an emergency fuel reserve, and is readily broken down when blood glucose levels drop. The breakdown of glycogen is known as glycogenolysis. Changes in enzymatic function within this carbohydrate metabolic pathway can lead to several diseases, such as diabetes and a multitude of glycogen storage diseases.
Nucleotides used in RNA synthesis are similar to those used in DNA synthesis, with the exception of thymidine — uracil is used in RNA synthesis. However, the removal of an -OH group on the ribose sugar is necessary to generate the deoxyribose sugar that makes up the nucleotide building blocks of DNA. Importantly, the breakdown of DNA and RNA is happening continuously in the cell. The purine and pyrimidine products of DNA and RNA breakdown can either be recycled for future nucleic acid synthesis or removed as waste products. The cyclical breakdown and synthesis of the various types of nucleic acids is critical for energy storage and generation and is, therefore, essential for cellular homeostasis.
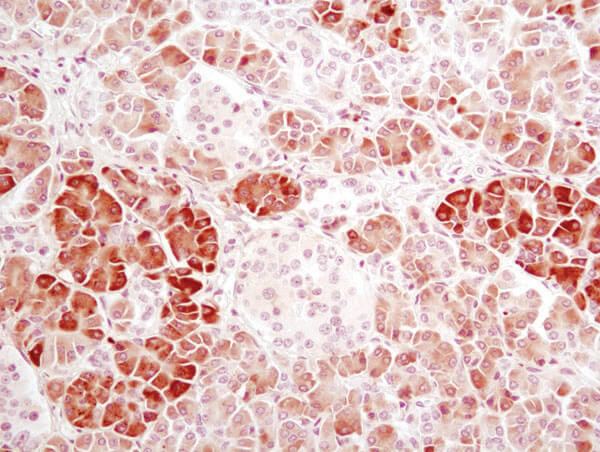
α-Amylase (D55H10) XP® Rabbit mAb #3796: IHC analysis of paraffin-embedded human pancreas using α-Amylase (D55H10) XP® Rabbit mAb #3796.
Amino Acid Metabolism
Proteins serve many functions: providing intracellular and extracellular structure (cells/tissues/organs); signaling; transporting cargo; acting as enzymes (e.g., protein catalyst); and providing immunity. Proteins are composed of amino acids linked in series to form polypeptide chains which, upon three-dimensional folding, form the mature protein. Because proteins constitute a significant portion of all cells, they are also a key nutrient that cells use for fuel and other processes.
Digestion of proteins begins in the stomach where stomach acid and the enzyme pepsin break down proteins into simpler polypeptides. These polypeptides are then further broken down into their constitutive amino acid building blocks that can either be recycled for formation of other proteins or further broken down by the liver into α-keto acids that can be utilized for energy, glucose, fat, or new amino acid formation. Amino acid breakdown can lead to the production of ammonium ions. Below, we discuss the urea cycle and its role in clearing toxic ammonia. It is important to note that proteins are a very useful source of energy during times of starvation. This is because the breakdown of proteins leads to the creation of metabolic intermediates that can feed into the citric acid cycle (discussed below).
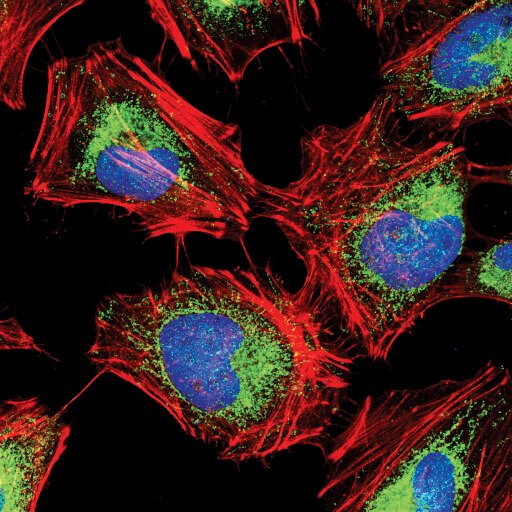
Hexokinase I (C35C4) Rabbit mAb #2024: Confocal IF analysis of HeLa cells using Hexokinase I (C35C4) Rabbit mAb #2024 (green). Actin filaments have been labeled with DY-554 phalloidin #13054 (red). Blue pseudocolor = DRAQ5 #4084 (fluorescent DNA dye).
Nucleotide Metabolism
Nucleotides fall into two major groups: purines and pyrimidines, both of which are composed of a phosphate group and a pentose sugar but differ in the size of their nitrogenous base.
Adenine and guanine are purines, while cytidine, uridine, and thymidine are pyrimidines. Purine and pyrimidine synthesis requires the input of energy in the form of adenosine triphosphate (ATP) and/or guanosine triphosphate (GTP). ATP and GTP are the primary energy carriers of the cell and contain high levels of energy in the bonds between their phosphate groups. The breaking of one of the bonds leads to the release of energy that can drive cellular functions. Phosphate groups can be sequentially removed via phosphatase enzymes from ATP and GTP to generate adenosine and guanosine di- and mono-phosphates (i.e., ADP, GDP, AMP, GMP). This is a reversible reaction, and phosphate groups can be added onto ADP/GDP and AMP/GMP by a group of enzymes known as kinases. Within the pyrimidine synthesis pathway, there are also UTP and CTP nucleotides, which are derived from uridine and cytidine, respectively.
Mitochondria and Mitochondrial Function in Metabolism

Mitochondria: Mitochondrial landscape.
Mitochondria are intracellular organelles that play a central role in cellular metabolism and energetics. These dynamic organelles play critical roles for integrating different metabolic inputs, compartmentalizing key metabolic and cell fate pathways, and acting as the efficient engine that converts nutrient inputs into energy currency, mainly ATP. Generating ATP from nutrient metabolites is seen as the key aspect of mitochondrial function. The conversion of metabolites from the citric acid cycle into ATP is achieved by the transfer of electrons from these intermediates through the respiratory chain subunits on the mitochondrial inner membrane. As electrons are transferred, protons are pumped into the inner membrane space, which establishes a biochemical gradient. The key respiratory complexes I, III, and IV reside in the inner mitochondrial membrane and are the protein units that pump the protons from the matrix into the inner membrane space using electrochemical energy derived from the transfer of electrons. Molecular oxygen is the final electron acceptor from complex IV, resulting in its reduction to H2O. Importantly, the imbalance of protons between the mitochondrial matrix and inner membrane space creates an electrochemical gradient, which provides the potential energy to drive ATP synthesis from ADP. This efficient energy-generating process, termed "aerobic respiration" since it depends on oxygen, yields the best energy return (ATP) on the initial nutrient input compared to other generating processes in the cell (such as anaerobic respiration, which does not require oxygen and produces smaller amounts of ATP; this occurs mainly from glycolysis and conversion of glucose to lactic acid). Mitochondrial function and energy generation are especially critical in energy-demanding tissues and cells of the body, such cells in the brain (neurons), heart (cardiomyocytes), and pancreas (beta-islets). Mitochondrial function is critical for all cells, and disease results when mitochondrial function is compromised. The severity of the resulting disease depends on the metabolic demands of the affected cell type(s). The different metabolic disorders that can arise from mitochondrial dysfunction are reviewed in Section 4.
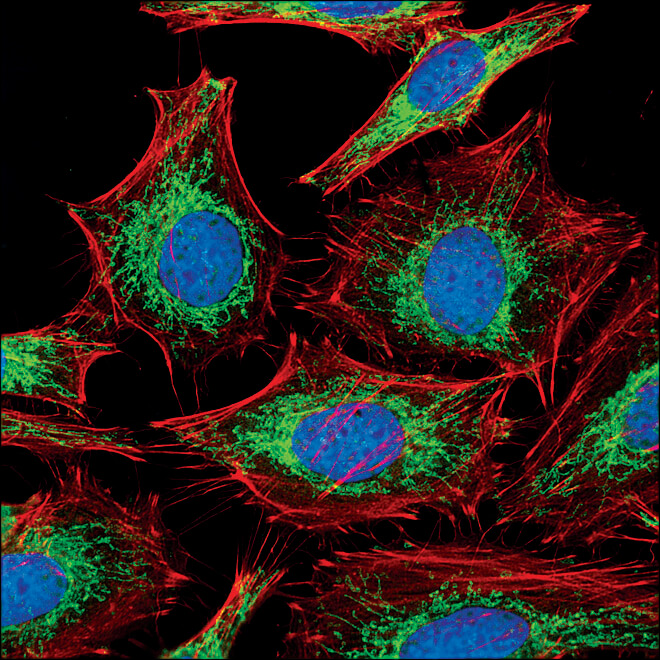
COX IV (3E11) Rabbit mAb #4850: Confocal IF analysis of HeLa cells labeled with COX IV (3E11) Rabbit mAb #4850 (green). Actin filaments have been labeled with Alexa Fluor® 555 phalloidin #8953 (red). Blue pseudocolor = DRAQ5 #4084 (fluorescent DNA dye).
Mitochondria are also known to have a unique physiology that works to regulate their function and impact on cellular metabolism. Mitochondria do not exist as single entities in the cell but instead form a dynamic network throughout the cytoplasm. The life cycle of a mitochondrion within the network is defined by the processes of biogenesis, fusion and fission, motility, and degradation. Biogenesis is the formation of new mitochondria. Fusion is when separate/distinct mitochondria merge together to become one; fission is the reverse: a single mitochondrion splitting into separate/distinct mitochondria. These processes are continuously happening as mitochondria respond to the variable energetic demands of the cell, and as a response to mitochondrial damage, in order to preserve cellular mitochondrial function. Mitochondrial movement or motility allows mitochondria to move to where their particular functions are needed. Finally, mitophagy is an autophagy-like process specifically affecting mitochondria which allows for degradation of mitochondrial components. Autophagy especially allows for the removal of damaged mitochondria. Defective mitochondrial autophagy, or mitophagy, leads to buildup of dysfunctional mitochondria. Defects in mitophagy have been implicated in the pathogenesis of some diseases, notably Parkinson's disease. Thus, proper regulation and maintenance of mitochondrial function is a critical aspect for the maintenance of metabolic homeostasis.
Major Metabolic Pathways
Below, we discuss the major metabolic pathways used by cells. Each pathway relies on enzymes to catalyze the specific chemical reactions involved in converting an original metabolic substrate into another through multiple intermediate steps that each produce metabolites.
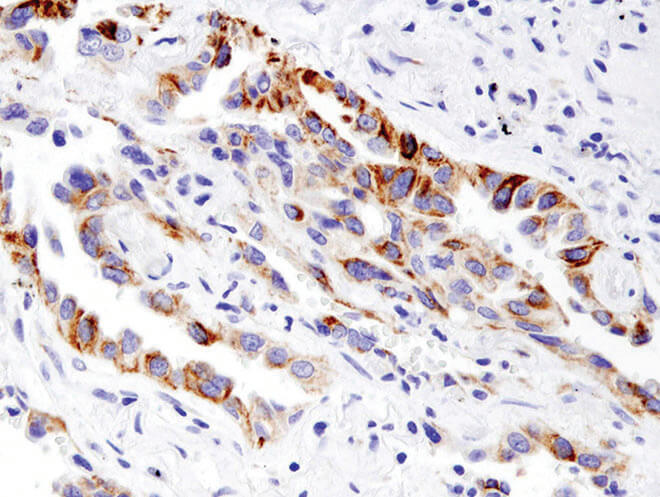
BNIP3 (D7U1T) Rabbit mAb #44060: IHC analysis of paraffin-embedded human non-small cell lung carcinoma using BNIP3 (D7U1T) Rabbit mAb #44060.
Anabolic vs Catabolic vs Amphibolic Metabolism
Macrophages can be triggered to recognize antigens, such as damaged cells or foreign material, for on-demand destruction. Macrophages are present in most tissues and respond when needed to infections and dying cells. The recognized material is destroyed via phagocytosis in the macrophage, which gives the cells their name ("big eater" in Greek). Macrophages take various forms when present in different locations and can perform additional functions besides phagocytosis.
Upon tissue injury or pathogen infection, monocytes in the blood will be recruited to the affected tissue and differentiate to make macrophages. Depending on the tissue localization, different types of macrophages exist, such as Kupffer cells in the liver, alveolar macrophages in the lung, microglia in the brain, etc. These different types of macrophages all come from monocytes but specialize their function to the resident tissue. Most of the general phagocytosis function is carried out by resident tissue macrophages. Besides phagocytosing dead cells and foreign material, macrophages can also signal to other immune cells via cytokines. To a certain extent, macrophages perform the critical function of antigen presentation, accordingly working together with T cells to support adaptive immunity. Additionally, macrophages can secrete cytokines such as IL-12 and play a role in local immune responses, while others secrete high amounts of IL-10, which mediates their role in tissue repair. Thus the "big eaters" play a variety of roles in the immune system in addition to the main job of phagocytosis.
Glycolysis
Glycolysis refers to the intracellular breakdown of glucose into pyruvic acid and ATP. Glycolysis takes place in the cytoplasm and utilizes a series of enzymes to split each six-carbon glucose molecule into two three-carbon pyruvate molecules. Glycolysis does not require oxygen and is, therefore, the predominant catabolic pathway in anaerobic organisms. Glycolysis is also utilized by "otherwise aerobic" organisms when oxygen is limited.
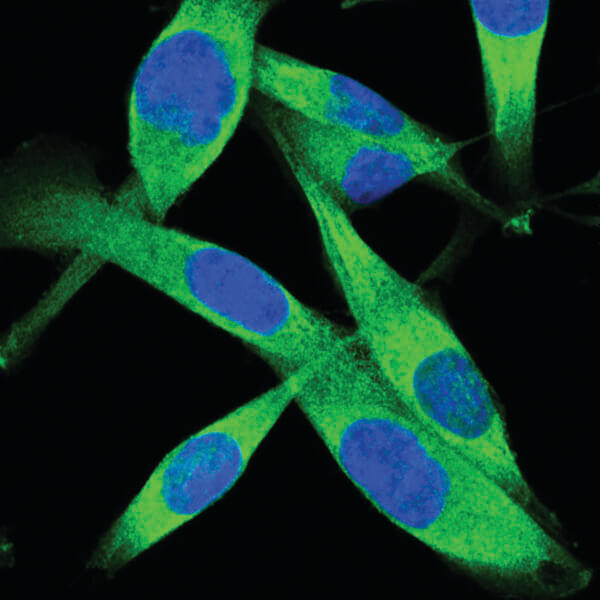
PKM2 (D78A4) XP® Rabbit mAb #4053: Confocal IF analysis of A204 cells using PKM2 (D78A4) XP® Rabbit mAb #4053 (green). Blue pseudocolor = DRAQ5 #4084 (fluorescent DNA dye).
Citric Acid Cycle (Krebs Cycle)

Krebs Cycle: Proteins of the Krebs cycle.
The citric acid cycle, also known as the Krebs cycle, is the next step in intracellular glucose metabolism. It takes place in the mitochondria and is initiated by acetyl coenzyme A (acetyl CoA), which is an oxidized derivative of the pyruvate produced during glycolysis. The citric acid cycle requires oxygen and releases carbon dioxide and water as byproducts. It is composed of a series of redox reactions that generate the high-energy molecules NADH, FADH2, and ATP. For each glucose molecule, two pyruvates are produced via glycolysis; thus, the citric acid cycle goes around twice and produces two carbon dioxide molecules, three NADH, one FADH2, and one ATP for each turn. While the citric acid cycle does not itself produce much ATP, the major energy currency of the cell, the NADH and FADH2 molecules act as electron carriers that shuttle into the electron transport chain for oxidative phosphorylation and high-energy production.
Oxidative Phosphorylation
The final stage of cellular respiration is oxidative phosphorylation (OXPHOS). OXPHOS also takes place in the mitochondria, across the inner membrane. There are five transmembrane enzyme complexes that drive the transfer of electrons from one molecule to another in a series of redox reactions. This is known as the electron transport chain. The transfer of electrons along the electron transport chain releases energy that then drives proton pumps to translocate protons against their concentration gradient from the mitochondrial matrix, across the inner mitochondrial membrane, and into the intermembrane space. This is the origin of the electrochemical gradient the drives OXPHOS. Accumulated protons in the intermembrane space then pass through the final complex in the electron transport chain, ATP synthase, along their concentration gradient into the mitochondrial matrix. The energy released by protons flowing down their concentration gradient through the ATP synthase molecule drives its function as a "molecular motor" that uses the energy to catalyze the addition of a phosphate group to the ADP precursor, creating ATP. One glucose molecule yields 30 to 36 ATP molecules from oxidative phosphorylation.
Pentose Phosphate Pathway
As discussed above, in cellular respiration the pyruvate produced during glycolysis is shunted into the citric acid cycle and oxidative phosphorylation. However, there is an alternative cytosolic pathway that branches off glycolysis and leads to the formation of the sugars necessary for DNA and RNA production. The pentose phosphate pathway utilizes a molecule that is produced in the first step of glycolysis—glucose-6-phosphate. Glucose-6-phosphate is generated through the addition of a phosphate group to glucose and is used by the pentose phosphate pathway to generate NADPH, five-carbon sugars known as pentoses, and ribose 5-phosphate, which serve as precursor molecules for nucleotide synthesis. NADPH plays an important functional role in not only the pentose phosphate pathway but in other biosynthetic processes, including fatty acid metabolism and reactive oxygen species (ROS) control.
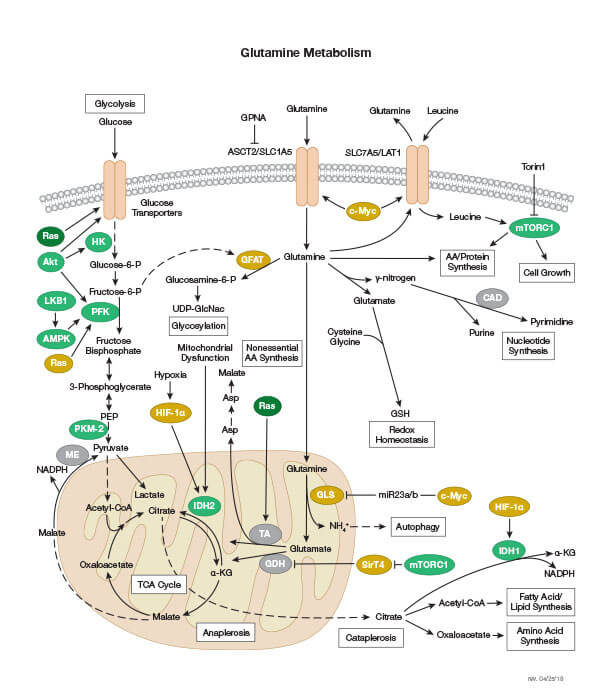
Glutamine Metabolism
Glutamine is an important fuel source in rapidly proliferating cells. It is transported into cells via a specific amino acid transporter and converted into glutamate in the mitochondria. There, the glutamate is converted into α-ketoglutarate, an intermediate in the citric acid cycle.
Urea Cycle
The urea cycle, also called the ornithine cycle, is necessary to prevent toxic buildup of ammonia in the body and occurs mostly in the liver. It is composed of biochemical reactions that produce urea from ammonium ions that are a byproduct of amino acid breakdown. In this cycle, carbon dioxide combines with the ammonia that is produced from the transamination of amino acids during protein metabolism, resulting in the generation of urea and water that are later eliminated as urine by the kidneys. The initial steps of the urea cycle take place in the mitochondria, and the later steps proceed in the cytosol.
Fatty Acid Synthesis
Fatty acids are both a source and storage unit of energy in the cell. In addition, fatty acids play a major role in cellular signaling and, therefore, heavily influence cellular function. Fatty acid synthesis takes place in the cytosol and involves the creation of fatty acids from acetyl-CoA and NADPH in a process that is catalyzed by fatty acid synthases. Acetyl-CoA units that are necessary for fatty acid synthesis are provided by the breakdown of glucose through glycolysis. Glucose breakdown also generates glycerol, which can combine with three fatty acid subunits to form triglycerides. Generation of phospholipids is also a critical part of fatty acid metabolism, as phospholipids are a major component of biological membranes. When glycerol combines with only two fatty acids and a phosphate group, this results in the formation of phospholipids. Phospholipids have many functions within the cell, but, most importantly, they form the lipid bilayer that makes up the cell membrane.
Aside from being the building blocks of cell and organelle membranes, phospholipids have been used in drug synthesis to increase permeability across the membrane and to improve drug bioavailability.
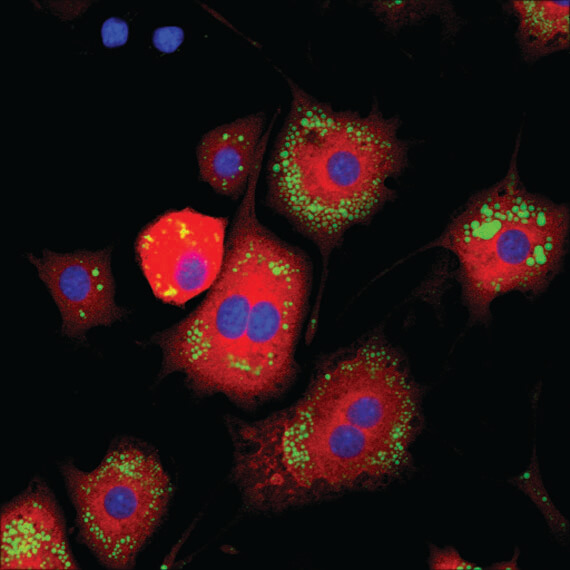
Lipin 1 (D2W9G) Rabbit mAb #14906: Confocal IF analysis of 3T3-L1 adipocytes (differentiated, 7 days) using Lipin 1 (D2W9G) Rabbit mAb #14906 (red). Lipid droplets were labeled with BODIPY® 493/503 (green). Blue pseudocolor = DRAQ5 #4084 (fluorescent DNA dye).
Fatty Acid Beta-Oxidation
Fatty acid beta-oxidation is the process by which fatty acids are broken down into their constituent acetyl-CoA subunits in the mitochondria. This acetyl-CoA then enters the citric acid cycle for sequential oxidation and the generation of NADH and FADH2.
Gluconeogenesis
Gluconeogenesis refers to the generation of glucose from sources other than carbohydrates. Similar to glycogenolysis, gluconeogenesis is an adaptive process that occurs primarily in the liver to ensure that blood glucose levels do not drop too low. Gluconeogenesis usually occurs during periods of low nutrient intake, intense exercise, or low-carbohydrate food consumption.
One-Carbon Metabolism
One-carbon metabolism refers to a group of folate-dependent metabolic pathways that are essential for the anabolism of several molecules, such as amino acids and nucleotides. In this pathway, folic acid acts as a carrier of one-carbon groups, facilitating the removal and transfer of these groups from donor molecules. There are three molecules that can be used to move one-carbon groups: tetrahydrofolate, a folate-derivative that acts as cofactor for several enzymes; s-adenosylmethionine, a methyl donor; and vitamin B12, a coenzyme in methylation and carbon rearrangement reactions. Aside from its role in amino acid and nucleotide synthesis, one-carbon metabolism is also important for DNA and histone methylation.
Oxidative Stress and Its Role in Metabolism
In mitochondria, the by-products of ATP generation via electron transport include reactive oxygen species (ROS), which are highly reactive molecules that can cause oxidative damage to organelles and other cellular structures. At low physiologic levels, ROS toxicity can be adequately controlled by intracellular antioxidant systems, including superoxide dismutase (SOD), glutathione, and catalase. Basal ROS levels are also now known to play a critical role in physiological cell pathways. However, pathological ROS levels have been shown to damage proteins, lipids, and DNA, which can lead to defective mitochondrial metabolism and deleterious consequences for cell function and viability. Effectively coping with ROS-induced mitochondrial damage is key to maintaining cell function and viability under different forms of cellular stress.
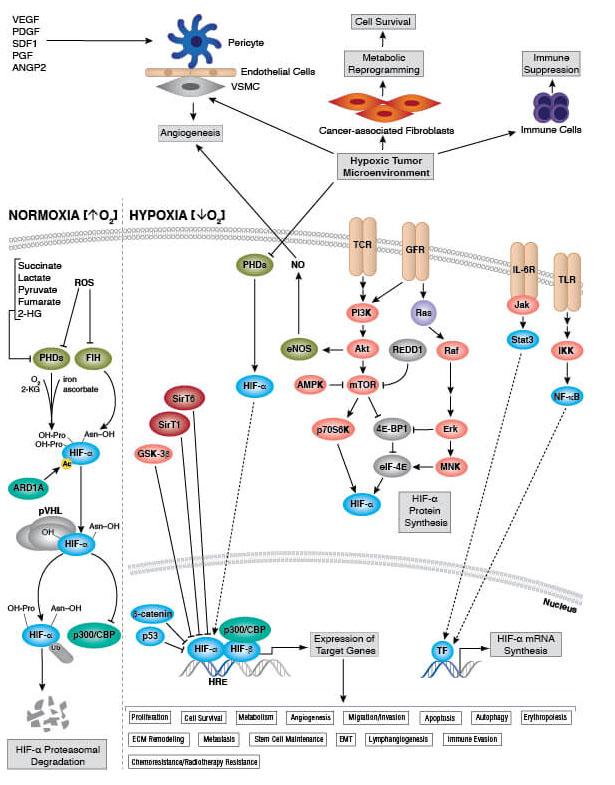
What Is Oxidative Stress
Different environmental conditions or cellular perturbations can induce oxidative stress. In the metabolic context, oxidative stress can occur when nutrient supply exceeds energy demand. This leads to a backup in the electron transport chain that can cause electrons to "leak" and react with O2 to form ROS. Dysfunction of respiratory chain components can also result in perturbed electron transport and increased ROS levels. Likewise, a deficit in antioxidant enzyme capacity, either due to decreased protein expression or decreased function, will allow ROS to accumulate. Though mitochondria are a major source of ROS, oxidative stress can also result from increased levels of ROS and reactive nitrogen species (RNS) from other cellular sources, including xanthine oxidase and cytochrome P450 oxidase systems in cells such as phagocytes.
Oxygen Species (ROS)
ROS come in different forms, with the two main groups being free radicals (species with unpaired electrons) and nonradicals (no unpaired electrons). When free electrons first react with O2, this forms superoxide anion (O2-), which is a very reactive but unstable ROS. Superoxide is quickly converted to hydrogen peroxide (H2O2) by superoxide dismutase (SOD). Though hydrogen peroxide is more stable, it can be converted to harmful hydroxyl radicals (-OH) following interactions with transition metals, in a process known as the Fenton reaction. Hydroxyl radicals are the most highly reactive ROS and, therefore, the most likely to cause oxidative damage to intracellular proteins and lipids.
Hypoxia and Cellular Respiration
Under hypoxic (oxygen-limiting) conditions, electron transport proceeds normally, but there is limited oxygen available to serve as final electron acceptors. In these cases, electron transport proceeds normally, but with no oxygen to accept the electrons. Left unchecked, this leads to increased electron leakage and ROS production. Accordingly, the cell has evolved specialized hypoxia response pathways that influence cellular respiration and associated oxidative stress. These pathways function to downregulate metabolic activity to avoid overwhelming the bioenergetic machinery. A key transcription factor, called hypoxia-inducible factor-1 (HIF-1), responds to reduced oxygen availability by decreasing electron transport chain activity and downregulating protein translation (an ATP-demanding process) and activity of Na-K-ATPase. This coordinated response allows the cell and mitochondria to withstand the period of lower oxygen until normal levels are restored.
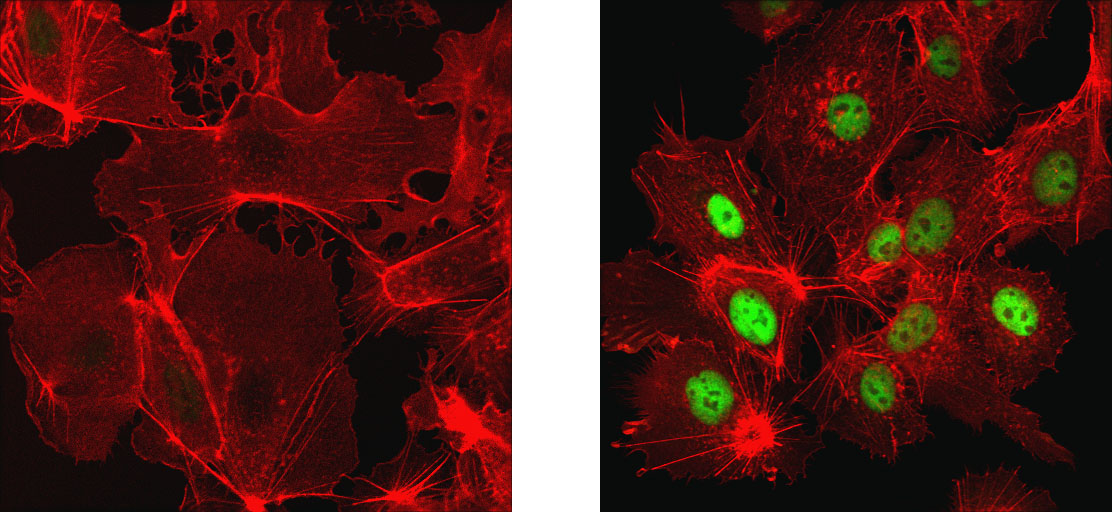
HIF-1α (D1S7W) XP® Rabbit mAb #36169: Confocal IF analysis of Hep G2 cells, untreated (left) or treated with cobalt chloride (500 μM, 24 h; right), using HIF-1α (D1S7W) XP® Rabbit mAb #36169 (green). Actin filaments were labeled with DyLight™ 554 Phalloidin #13054 (red).
Biomarkers of Oxidative Stress
Since oxidative stress is a multifaceted and complex process, defining the best biomarkers of oxidative stress in cells and tissues is challenging.
One approach is to measure biochemical modifications of target molecules (e.g., proteins) that occur as a result of oxidative damage — for example, protein carbonylation assays using 2,4-dinitrophenylhydrazine (DNPH) to detect protein products that have been chemically modified by reactive oxygen. Oxidized low-density lipoprotein is also a common biomarker, especially in cardiovascular disease. Lipid modifications can also be detected following oxidative damage. A common biomarker is 4-hydroxynonenal (4-HNE), a hydroxyalkenal produced in response to lipid peroxidation. One may also measure biochemical changes in endogenous antioxidant system activities, such as glutathione levels, as indications of oxidative stress. DNA and RNA are also targets of ROS, such that oxidation of these nucleotides can be used as biomarkers of oxidative stress. Researchers may also look at perturbations to common pathways known to be regulated by oxidative stress, such as Nrf2 and HIF-1. The search for robust biomarkers of oxidative stress is continually evolving, so one should review the recent literature to determine what assays best suit their needs.
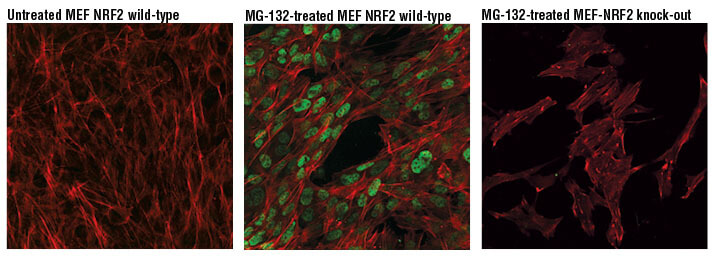
NRF2 (D9J1B) Rat mAb (IF Specific) #14596: Confocal IF analysis of MEF NRF2 wild-type cells, untreated (left) or treated with MG-132 #2194 (10 μM, 8 hr; center) and MEF NRF2 knock-out cells treated with MG-132 #2194 (10 μM, 8 hr; right), using NRF2 (D9J1B) Rat mAb (IF Specific) #14596 (green pseudocolor). Actin filaments were labeled with Alexa Fluor® 488 Phalloidin #8878 (red pseudocolor).
Metabolic Disorders
Due to the critical importance of maintaining cellular metabolic homeostasis, disruptions to metabolic pathways can lead to a plethora of metabolic disorders. Below, we summarize major metabolic disorders and their underlying causes.
Causes of Metabolic Disorders
Metabolic disorders can occur as a result of genetic mutations or in response to environmental perturbations that functionally affect major digestive organs or those that serve important metabolic functions (e.g., liver, pancreas).
Types of Metabolic Disorders
Currently, the National Institutes of Health classify metabolic disorders into the following classes:
- Acid-base imbalance
- Metabolic brain diseases
- Disorders of calcium metabolism
- DNA repair-deficiency disorders
- Glucose metabolism disorders
- Hyperlactatemia
- Iron metabolism disorders
- Lipid metabolism disorders
- Malabsorption syndromes
- Metabolic syndrome X
- Inborn error of metabolism
- Mitochondrial diseases
- Phosphorus metabolism disorders
- Porphyrias
- Proteostasis deficiencies
- Metabolic skin diseases
- Wasting syndromes
- Water-electrolyte imbalance
Prominent Metabolic Disorders
There are prominent metabolic diseases that constitute the majority of the metabolic disorders that affect the population, namely diabetes/insulin resistance, obesity/metabolic syndrome, cardiovascular disease, renal tissue injury, and inborn errors of metabolism.
Cancer and Metabolism
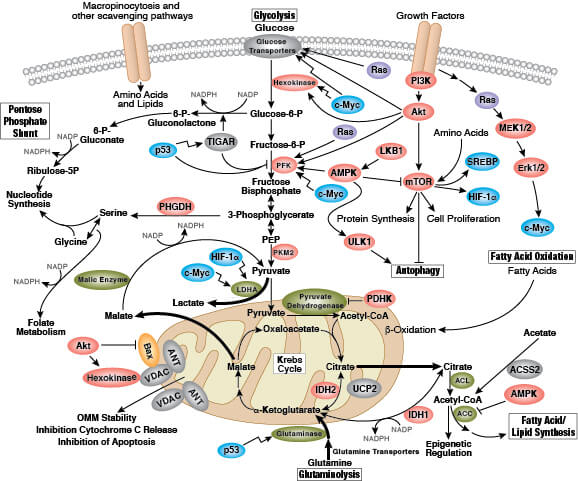
Metabolism is an increasingly important aspect of cancer cell biology. Early insight into the role of metabolism in cancer research was due to findings by Otto Warburg that showed cancer cells rely on the inefficient metabolic pathway of fermentation rather than the much more energy efficient aerobic respiration. This "Warburg effect" is a hallmark of many cancers that reflects a metabolic adaptation by the cancer cells that promotes their growth and survival. The prevailing hypothesis is that these metabolic adaptations provide a survival advantage to cancer cells over normal cells, since robust generation of cellular building blocks (i.e., proteins, lipids, nucleic acids) in oxygen-limited conditions is required to support their rapid, uncontrolled proliferation. Another line of research suggests that cancer cells need to use their nutrient inputs as cellular building blocks; thus, they switch from catabolic (nutrient consuming) to anabolic (biomass building) pathways to support their high proliferation rates. In addition to the Warburg effect, there are other documented hallmarks of cancer metabolism, including impaired uptake of glucose and amino acids, utilization of different modes of nutrient acquisition, harnessing glycolysis/TCA cycle intermediates for biosynthesis and NADPH production, higher demand for nitrogen, changes to metabolite-driven gene regulation, and metabolic interactions with the microenvironment.
Diabetes/Insulin Resistance
Diabetes occurs when blood glucose levels are consistently elevated above physiologically healthy levels. There are two types of diabetes, type 1 diabetes and type 2 diabetes. In type 1 diabetes, which manifests typically in childhood or early adulthood (which could include adolescence), the body fails to produce sufficient insulin, the hormone that induces glucose uptake from the blood into cells. Diabetes is a metabolic disease that results from autoimmune destruction of beta cells in the pancreatic islets, the insulin-making cells of the body. In type 2 diabetes, insulin is either not made or the body has been desensitized to the presence of insulin (i.e., insulin resistance). Type 2 diabetes can manifest at any age and is typically the result of unhealthy diet. People with type 1 and type 2 diabetes are at a much higher risk of developing heart disease, kidney disease, dental disease, circulation abnormalities, eye disease, and nerve damage.
Obesity
Obesity is an excess of adipose tissue, which may be stored subcutaneously or as visceral fat. Obesity is typically diagnosed using the body-mass index, a calculated value that represents body weight as a function of height, scaled according to sex and age. Obesity is one of the most prevalent diseases in the United States, with staggering statistics that are rising annually. Obesity is strongly associated with other health complications, including heart and cardiovascular disease, type 2 diabetes, and even some cancers and is one of the leading causes of premature, preventable death. While some people are genetically predisposed to obesity, most obesity cases arise because of diet and associated lifestyle choices.
Metabolic Syndrome
Metabolic syndrome is another disorder that is linked to obesity. It is a cluster of metabolic disorders that occur in combination and which are also strongly associated with other health complications, such as diabetes, heart disease, and stroke. A diagnosis of metabolic syndrome is given when a patient presents with three of the following conditions: abdominal obesity, elevated serum triglyceride, low serum HDL (good cholesterol), high blood pressure, and high fasting blood glucose levels.
Cardiovascular Disease
The American Heart Association defines cardiovascular disease as one or more of the following conditions: heart disease, heart attack, stroke, heart failure, arrhythmia, and heart valve problems. Obesity and diabetes greatly increase the risk of cardiovascular disease. In addition, because of a high reliance on energy, cardiovascular disease can occur independent of metabolic syndrome/obesity when cardiac cells have disrupted intracellular metabolic pathways.
Renal Tissue Injury and Function
Metabolic syndrome is also associated with chronic kidney (renal) disease. Different clinical features are associated with metabolic syndrome and thought to contribute to renal dysfunction and tissue injury. These include hyperinsulinemia, activation of the renin-angiotensin-aldosterone system, increased oxidative stress, and inflammatory cytokine production.
Inborn Errors of Metabolism
Inborn errors of metabolism are rare genetic diseases that lead to the dysfunction or absence of key enzymes involved in metabolic pathways. Below, we discuss lysosomal storage diseases, amino acid metabolism disorders, and carbohydrate metabolism disorders.
Lysosomal Storage Diseases
Lysosomal storage diseases are a collection of diseases in which the lysosome is deficient in key lysosomal enzymes, thereby decreasing breakdown and increasing storage of lysosomal material targeted for degradation. Two examples of lysosomal storage disease are Gaucher disease and Niemann-Pick disease. Gaucher disease results from the deficiency of the enzyme glucocerebrosidase and leads to the buildup of glucocerebroside, a sphingolipid, in white blood cells and macrophages. Niemann-Pick disease is a lipid storage disorder in which pathological levels of lipids accumulate in the spleen, liver, lungs, bone marrow, and brain. There are four types of Niemann-Pick disease that are classified based on their genetic basis and the associated symptoms.
Amino Acid Metabolism Disorders
Amino acid metabolism disorders are genetic diseases that affect amino acid catabolism. Two examples of amino acid metabolism disorders are phenylketonuria and maple syrup urine disease. Phenylketonuria occurs in infants that cannot break down the amino acid phenylalanine into tyrosine. This leads to the buildup of phenylalanine in the blood. Phenylalanine is toxic to the brain, and if phenylketonuria is left untreated, it can cause developmental defects that result in intellectual disability. Similarly, maple syrup urine disease occurs in children who are unable to break down leucine, isoleucine, and valine leading to the buildup of amino acid byproducts, causing body fluids to smell like maple syrup, and eventually leading to neurologic dysfunction.
Carbohydrate Metabolism Disorders
Carbohydrate metabolism disorders result from the dysfunction of enzymes involved in carbohydrate metabolism. One subset of disorders, known as glycogen storage diseases, are inherited disorders that result from mutations to key enzymes involved in the biochemical processing of glycogen. There are more than 10 types of glycogen storage diseases that are classified based on the enzyme that is affected. The organ system that is affected depends on the tissue distribution of the affected enzyme, therefore a wide array of symptoms can result. Another example of a carbohydrate metabolism disorder is glycogen-6-phosphate dehydrogenase (G6PD) deficiency, which is involved in the catabolism of carbohydrates. This G6PD deficiency leads to the lysis of red blood cells because of a buildup of reactive oxygen species, thereby leading to symptoms of hemolytic anemia.